Pain is determined by the neurologic properties of receptor organs, neurons, and their interconnections. These may become supersensitive or hyperactive following denervation (Cannon's Law). A common cause of denervation in the peripheral nervous system is neuropathy or radiculopathy as a sequel to spondylosis. Spondylosis in its early stage may be "asymptomatic" or painless and hence unsuspected, because small-diameter pain fibers may not initially be involved despite the attenuation of the other component fibers of the nerve. The term "prespondylosis" is introduced here to describe this presently unrecognized phase of insidious attrition to the other functions of the nerve, especially the trophic aspect. It is postulated that many diverse pain syndromes of apparently unrelated causation may be attributed to abnormal noxious input into the central nervous system from supersensitive receptor organs (nociceptors) and hyperactive control systems at internuncial pools. Furthermore, trauma to a healthy nerve is usually painless or only briefly painful, unless there is preexisting neuropathy. Some pain syndromes in muscle (eg, trigger points and myofascial pain syndromes) and nerve (eg, causalgia and diabetic neuropathy) that may be related to denervation are discussed. [Key words: spondylosis, denervation, hypersensitivity, pain syndrome].
Pain is merely an emotional response to afferent input; its perception is obviously influenced by emotion and dependent upon personality and mood. It is not a sensation in the strict neurophysiologic sense since there is no direct relation between the intensity of the applied stimulus and the intensity of evoked experience. Yet, however complex the phenomenon of pain may appear to be, the flow of events from input of information into the nervous system (whether it be from a noxious or other stimulus) to final evoked response is determined by the neurobiologic properties of neurons and their interconnections. All forms of adequate stimuli, both from the external world and from within the body, activate receptor organs. The information gathered by these receptor organs is transmitted to the central nervous system by way of primary afferent fibers. These synapse either directly on motoneurons or, more commonly, on interneurons. The latter may activate other interneurons in either the spinal cord or the brain. The patterns of interaction among these cells can be exceedingly complex. Eventually, however, the interneuron chains feed information to motoneurons, and these in turn command actions by effectors which include muscle and gland cells (see Figure 1).
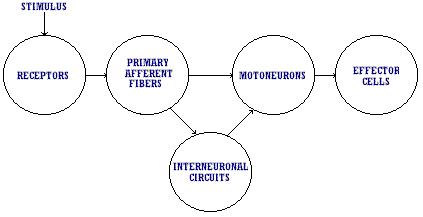
This paper draws attention to the important but neglected role of supersensitivity of denervated structures in the possible modification of afferent inputs and internuncial circuits. It is postulated that many diverse pain syndromes of apparently unrelated causation can probably be attributed to "denervation supersensitivity" and /or hyperactive control systems at internuncial pools. The concept of "prespondylosis," or the early pain-free stage of spondylosis, as a cause of unsuspected peripheral neuropathy and denervation supersensitivity is introduced.
Among the mysterious phenomena handed on from the physiologists of the 19th century to those of this century were two that were subsequently shown to have a common basis: the "paradoxical pupillary dilation" and the "Philipeaux-Vulpian" or "pseudomotor phenomenon."
It had first been noticed in 1855 that in an experimental animal, severance of the left cervical sympathetic nerve (preganglionic fibers) and simultaneous severance of the sympathetic branches above the right superior ganglion (postganglionic fibers) was followed by a curious difference in the two eyes: after approximately 48 hours the right pupil was larger than the left. Both irises had been deprived of their sympathetic connections, but the right pupil, deprived of its ultimate sympathetic nerve supply, was larger than the left, which was deprived of its penultimate supply.
The Philipeaux-Vulpian phenomenon described the anomalous response of denervated striated muscle to stimulation of non-motor nerves distributed to adjacent blood vessels. It was noticed that when the hypoglossal nerve (motor nerve of the tongue muscles) was severed and allowed to degenerate, stimulation of the chorda tympani (sensory, vasodilator, and secretory fibers, but no motor fibers) caused the tongue to contract mysteriously.
It was not until many decades later that the explanation for these two mysteries was traced to the increased sensitivity of denervated structures to circulating transmitter agents. Denervation, it was shown, sensitized the reactor muscle of the iris to circulating adrenaline, causing the paradoxic exaggerated retraction on the denervated side. The pseudomotor phenomenon in the tongue occurred when the muscles, following denervation supersensitivity, responded to acetylcholine liberated at the terminals of the vasodilator nerve. Most of the early research was by Cannon and Rosenblueth, who proposed a law of denervation (Cannon's Law), which stated, "When in a series of efferent neurons a unit is destroyed, an increased irritability to chemical agents develops in the isolated structure or structures, the effects being maximal in the party directly denervated." They showed that denervated striated muscle, smooth muscle, salivary glands, sudorific glands, autonomic ganglion cells, spinal neurons, and even neurons within the cortex develop supersensitivity. Today, repeated animal experiments have confirmed that denervation supersensitivity is indeed a general phenomenon. For example, in muscle, both striated and smooth, it has now been shown that there is an increase in the surface area of the muscle fiber that is sensitive to acetylcholine. Normally, the area of receptor sensitivity is very sharply circumscribed, but when the muscle loses its motor innervation there is a marked increase in the degree to which extrajunctional membrane responds to the application of acetylcholine. This change is detectable within a matter of hours and reaches a maximum in about a week, by which time the entire surface of the muscle fiber is as sensitive to acetylcholine as the normal end-plate region. This development of supersensitivity probably represents incorporation of newly synthesized receptors into extrajunctional membrane.
It is important to understand that actual physical interruption is not necessary for "denervation hypersensitivity" to develop. Minor degrees of damage or experimental exposure of motor axons to poisons such as colchicine or vinblastine can destroy the microtubules within the axons. Such a nerve still conducts nerve impulses, synthesizes and releases transmitted substances, and evokes both muscle action potentials and muscle contraction, but the entire membrane of the muscle cells innervated by the affected axon becomes supersensitive to the transmitter as if the muscle had been denervated. Destruction of microtubules within the axons is thought to disrupt axoplasmic flow and interfere with the trophic function of the nerve.
A second important change in muscle is the onset of spontaneous electrical activity of fibrillation. An innervated mammalian skeletal muscle normally gives an action potential only in response to the release of the transmitter agent. In contrast, action potentials begin to occur spontaneously within a few days after denervation and continue for as long as the muscle remains denervated, in some cases up to a year or more. This autogenic activity probably arises from local fluctuation in membrane potential and from an increase in membrane conduction to electrolytes. Other changes include those in muscle structure and biochemistry. Muscle atrophy eventually occurs following a progressive destruction of the fiber's contractile elements, resulting in a decrease in fiber diameter and slowing the speed of the contractile response. Another important but little understood change of denervated muscle fibers is a renewed ability to receive synaptic contacts. Unlike normal muscle fibers which resist innervation from foreign nerves, denervated muscle fibers accept contacts from a wide variety of sources, including other motor nerves, preganglionic autonomic fibers, and possibly even sensory nerves.
There are similar changes in neurons, but neurons are generally more difficult to investigate than muscle fibers because neuronal innervation is usually widely distributed on the soma and dendrites. Much of the early work also came from Cannon and his fellow workers, but it was not until the recent application of differential interference contrast microscopy (which allows visualization of living neuronal synapses) that sensitivity to acetylcholine was shown to be encountered at every point on the cell surface instead of only at the normal synaptic regions. Other effects of denervation on neurons have yet to be studied, but spontaneous activity of denervated sympathetic nerves has been described and has been suggested to be analogous to the fibrillation of denervated muscle fibers. As in muscle fibers, denervation of neurons induces sprouting of nearby presynaptic elements, and nerve cells are more receptive to foreign innervation , with denervated autonomic neurons particularly prone to receive a variety of foreign synapses. Biochemical studies of peripheral neurons also show enzymatic changes following denervation, and it has been demonstrated that these too may affect the long-term regulatory mechanism in the peripheral and autonomic nervous systems.
Changes at synapses also occur. The studies of Hughes, Kosterlitz, and others have shown that endogenous morphine-like peptides (endorphins and enkephalins) inhibit neuronal activity by altering sodium conductance at opiate receptors in the brain and at the spinal cord levels. Methionine-enkephalin is a neurotransmitter found in spinal gray matter occurring at the terminals of interneurons. Excitation of these interneurons, which interact with one another and impinge on the nerve endings of sensory neurons, produces primary afferent depolarization or presynaptic inhibition and attenuates nociceptive transmission across the synapses of primary afferent fibers and second order neurons, especially in Laminae I, II, and III. Chronic lesions of the primary afferents decrease the number of opiate receptors in the dorsal horn with a corresponding reduction of interneuron activity and presynaptic inhibition by enkephalin. Peripheral nerve disease may therefore also cause facilitation of noxious inputs at the dorsal horn.
The simple idea of a closed chain of neurons producing an invariable response when stimulated is no longer tenable, yet the fundamental physiologic fact remains that once an action potential is initiated in a receptor organ by a threshold stimulus, it is propagated to the central nervous system by way of primary afferent neurons that synapse either directly on motoneurons or, more commonly, on interneurons. It is the pattern of interaction among interneurons and multineuronal assemblies in the spinal cord and in the brain that can be exceedingly complex, modifying the message on its way to the brain, possibly diverting it into other pathways or suppressing it completely.
Three basic concepts have been formulated to explain the peripheral encoding of painful stimuli. These are 1) intensity coding, 2) the pattern theory, and 3) the specificity hypothesis. Despite arguments to the contrary, the evidence is compelling that some receptors (nociceptors) and neurons are at least relatively specialized to signal stimuli of tissue-damaging intensity. However, because excitation of receptors other than nociceptors can contribute to the sensation of pain, a modified polymodal pattern concept has also been proposed. Nociceptors consist of the terminations of thinly myelinated Group A&(Group III) fibers, diameter 1-4 mm and conducting at 5-45 meter/sec ("fast pain"), and C (Group IV) non-myelinated fibers, which are thinner and conduct at about 1 meter/sec ("slow pain"). These fibers synapse with neurons in the dorsal horn and are relayed via interneurons to higher centers, probably with control systems to regulate the input of noxious stimuli at several levels. Because supersensitivity occurs as a general phenomenon following denervation, heightened neuronal and interneuronal activity may exist throughout the nervous systems - peripheral, central, and autonomic.
In the peripheral nervous system, a common cause of neuronal destruction is peripheral neuropathy when there is disordered function and/or structure of the peripheral nerve. While the causes of peripheral neuropathy are many and varied (congenital, neoplasm's, inflammatory, traumatic, vascular, toxic, metabolic, infective, degenerative, idiopathic, and others), the peripheral nerve responds with only a limited repertoire of pathologic reactions. This may be either attenuation of the caliber of axons or primary damage to myelin, but is usually a combination of both. Variable degrees of damage with variable degrees of reversibility may be present, ranging from neurapraxia to axonotmesis and neurotmesis. Peripheral neuropathy may occur at various sites, but the spinal root within the spinal canal and intervertebral foramina, and even after it emerges, is especially prone to damage. This may follow acute trauma, but more usually it is the long-term sequel of spondylosis which causes simultaneous damage to the nerve roots (radiculopathy) and cord (myelopathy). Spondylosis (which refers to the structural disintegration and morphologic alterations in the intervertebral disc and pathoanatomic changes in surrounding structures) has been acknowledged as a clinical entity only for some 20 years, although even today the significance of the silent, pain-free, but not necessarily morbidity-free, prespondylotic phase is still not widely recognized. "Prespondylosis" may be "symptomless," its symptoms and signs unsuspected, because pain may not be a feature. Pain occurs only when and if the degenerative changes impinge upon local pain-sensitive structures to produce local pain, or upon pain fibers of the nerve root to produce the transmitted pain of "radiculitis," a clinical term commonly used to describe the discomfort or pain radiating along the peripheral nerve. However, constant attrition of the peripheral nerve can attenuate fibers other than those of pain (which are small and less liable to mechanically caused ischemia), producing insidious neuropathy, the effects of which are projected onto the dermatomal, myotomal, and sclerotomal target structures supplied by the segmental nerve. Dysfunction may be motor, sensory, trophic, or autonomic, but since pain fibers are not necessarily involved, there are no "symptoms" and both patient and physician may be oblivious to the condition. "Prespondylosis" nonetheless has its implications and may contribute to chronic pain. For example, whereas acute structural deformation of a healthy nerve is not painful or only briefly so (eg, peroneal nerve palsy or radial nerve "Saturday night" palsy), such is not the case in an unhealthy nerve. It has recently been shown that when and if pain develops in a peripheral nerve, it is primarily associated with the acute breakdown of myelinated fibers (either Wallerian or axonal degeneration) superimposed on the preexistence of chronic nerve fiber degeneration. Pain is probably not caused simply by the different proportions of large to small fibers remaining after nerve degeneration as anticipated by the gate theory, but by the acute upon chronic or recent abnormal rate of breakdown of myelinated fibers (whatever is primary cause may be). Animal experiments have been furthermore shown that an acute mechanical injury to a healthy dorsal nerve root does not produce a sustained discharge unless there has been preexisting minor chronic injury to the nerve. Clinically, it is also common knowledge that in asymptomatic subjects the mere appearance of degenerative changes in spinal roentgenograms is not of much clinical significance, but in these persons, disability after injury will tend to be prolonged and signs of radiculopathy more commonly found. It would therefore appear that for pain to persist after trauma, a prerequisite is the existence of chronic nerve irritation.
Myalgic hyperalgesia, or excessive tenderness to digital pressure, is not a normal feature of muscle because their mechanosensitive nociceptors are located deep within the muscle bulk and have high thresholds. (Muscle A& fibers are mechanosensitive, have high thresholds, and respond to strong localized pressure but not to stretch or ischemia. Muscle C fibers also have high mechanical thresholds but in addition are excited by ischemia combined with contraction of the muscle.) Myalgic hyperalgesia may be local or traumatic following local injury and tissue damage when algogenic chemical substances such as 5-hydroxytrypatamine, histamine, bradykinin, and hydrogen ions are liberated. These produce an unspecific but powerful excitatory effect on noiceptors as well as on those low-threshold mechanoreceptors that have myelinated afferent fibers. Myalgic hyperalgesia may also be secondary to neuropathy when the noiceptors develop supersensitivity following denervation. Tenderness is then maximum at the neurovascular hilus where noiceptors are most abundant around the principal blood vessels and nerves as they enter the deep surface of the muscle to reach the muscle's motor zone of innervation. As this zone is fairly constant in position for each muscle, tenderness in muscles secondary to neuropathy is easily found. Tenderness at the muscle's zone if innervation is often loosely referred to as at the "motor point" (a point on the skin where a muscle twitch may be evoked in response to minimal electrical stimulation). Variable degrees of tenderness at motor points are usually present in the upper and lower limb muscles of persons who have some degree of spondylotic radiculopathy, the degree of myalgic hyperalgesia paralleling the radiculopathy. The presence of tenderness at motor points within an affected segmental myotome is therefore a useful diagnostic and prognostic aid following spinal injuries.
In some cases of denervation supersensitivity it may be possible for the afferent barrage from muscle nociceptors (at the zone of innervation and musculotendinous junctions) and their connections via spinal interneurons to become self-perpetuating, thus constituting, in effect, a "trigger zone or point." A comparison of the maps of trigger points produced by Travell and Rinzler with that of motor points will show their spatial coincidence. Furthermore, trigger zones may be demonstrated to coincide with motor points by electrical stimulation.
Many painful conditions that are presently labeled as vague clinical entities ("tendonitis," "bursitis," or "fibrositis") are probably hyperalgesic nociceptor regions in myofascial structures. For example, in midcervical spondylosis, tenderness at the anterior deltoid muscle motor point and the bicipital tendon is called "bicipital tendonitis." Tenderness at the wrist extensor muscle motor points and musculotendinous junctions around the lateral epicondyle of the elbow is commonly called "tennis elbow" or "lateral epicondylitis" (the tenderness at the bony epicondyle is probably sclerotomal). Myalgic hyperalgesia in the left pectoral muscles has been mistaken for angina and cardiac pain. "Bursitis" around the hip is not an uncommon diagnosis, yet surgical intervention rarely reveals a bursa distended with serous fluid. This "bursitis" is often tender gluteal muscle motor points secondary to lumbar spondylosis. These entities presently saddled with diverse, nondescript labels may be demonstrated by electrical stimulation to be motor points, and electromyography will generally show electrodiagnostic evidence of radiculopathy, but even simple palpitation can reveal hyperalgesia in the several muscles supplied by both anterior and posterior primary rami (ie, at root level) within the same segmental level or myotome. In these conditions, treatment should logically be addressed to the underlying spinal problem; in our experience, this has been followed by resolution of symptoms.
Supersensitivity of denervated structures may also lead to muscle spasm which is so often a co-feature of pain. Muscle tone may be increased at the muscle spindle whose intrafusal fibers, innervated from higher centers by the gamma motoneurons, may be subjected to increased impulse traffic. Hypersensitivity of the primary and secondary endings, which are sensitive to stretch of the central portion of the spindle, may also over-stimulate the essential feedback mechanism by which skeletal muscle and resting muscle tonus are controlled. The afferent discharge of the spindle via the dorsal root on the motoneurons of the same muscle is excitatory.
The extreme example of causalgia is discussed first, as its manifold manifestations represent all aspects of peripheral neuralgic hyperpathia. The term "causalgia" is derived from the Greek 'kausis', "burning," and 'algos' "pain," to describe the most striking feature of the condition, which is persistent, severe, and burning pain in an affected extremity, usually as the result of a partial injury to a nerve (commonly, the median, ulnar, and sciatic nerves). In addition to the pain there is invariably autonomic dysfunction and trophic changes in skin and/or bones in the involved part. Causalgic pain has been categorized as "major causalgia" and a less painful variant referred to as "minor causalgia" or "posttraumatic reflex sympathetic dystrophy." Typically, causalgic pain appears within a week following a nerve injury (when denervation supersensitivity has had time to develop), but its onset may be delayed by as much as three months. The severe, burning pain may be explained by hypersensitivity of receptors and small-diameter afferent fibers (A& and C) in cutaneous and other tissues. The autonomic dysfunction and trophic changes may likewise be the result of supersensitivity at lateral horn cells, autonomic ganglia, and receptors around blood vessels; thus, a sympathetic nerve block and/or sympathectomy provides relief in a proportion of patients.
Doupe and co-workers have suggested that trauma causes the formation of "artificial synapses" (ephapses) between sympathetic efferent and somatic sensory afferent nerves. According to this theory, a sympathetic impulse traveling down the efferent nerve, in addition to its usual effects, causes depolarization of the somatic sensory nerve at the point of artificial synapse. This depolarization is then propagated orthodromically along the afferent sensory impulses causes abnormally high sensory discharge when is felt as pain. In addition, depolarization at the artificial synapse is said to propagate antidromically along the somatic afferent, leading to the release of certain substances that decrease the threshold at the sensory nerve ending and further increase the impulses reaching central areas.
Livingston's theory of the "vicious cycle of reflexes" postulated that there is chronic irritation of a peripheral sensory nerve leading to increased afferent impulses and resulting in abnormal activity in an "internuncial pool" of neurons in the lateral and anterior horns of the spinal cord. The concept of denervation supersensitivity supports Livingston's theory, because peripheral receptors, afferent neurons, internuncial pools, and autonomic ganglia may become hypersensitive or hyperactive. Furthermore, autonomic neurons may generate spontaneous autogenic potentials similar to muscle fibrillations (see above). However, the increased receptivity of denervated autonomic neurons to a variety of foreign synapses and peripheral nociceptors to released algogenic substances also supports the theory of artificial synapses proposed by Doupe and co-workers. It is also significant that changes at spinal and other central synapses may occur (see above) with facilitation of noxious input.
In recent years the well-known gate theory of Melzack and Wall has been applied to causalgia (and to many other pain syndromes). It is suggested that cells in the substantial gelatinosa of the dorsal horn of the spinal cord acts as a "gate control system," modifying the transmission of afferent sensory impulses. This theory emphasizes a pattern of impulses rather than single impulses with a "selection process" to explain the intricacies of sensory experience. The gate theory contends that impulses from large myelinated fibers inhibit or "close the gate," whereas tonic background impulses transmitted along smaller fibers (which include afferent sympathetic fibers) "open the gate" to facilitate transmission. The theory also proposes a descending control system originating in the brain that modulates the excitability of afferent conduction. The "gate theory," published in 1965, was written before the present explosion of information about the anatomic state of nerves in the peripheral neuropathies. Wall and Melzack were influenced, in particular, by a study on postherpetic neuralgia in which it was shown that intercostal nerve biopsy specimens had a preferential loss of large myelinated fibers, and Noordenbos had generalized from this observation to propose that pain was a consequence of a loss of inhibition normally provided by the large fibers. It is now known that loss of large fibers is not necessarily followed by pain. In many conditions (eg, Friedreich's ataxia) there may be a large-fiber deficit without pain. Wall, now realizing that any attempt to correlate the remaining fiber diameter spectrum with pain is no longer possible, has restated the gate control theory of pain recently:
Therefore the brain receives messages about injury by way of a gate-controlled system that is influenced by 1) injury signals, 2) other types of afferent impulse, and 3) descending control.
In this restatement, Wall stated that fiber diameter alone is not enough or is even completely irrelevant to explain pain in the neuropathies when pathologic peripheral fibers have unusual impulse generation and conduction properties. However, the original proposal that transmission of information about injury from the periphery to the first central cells is under control (influenced by peripheral afferents and by descending impulses), still holds. In denervation supersensitivity, as mentioned above, facilitation of noxious input may occur at the "gate" in the dorsal horn from a reduction of presynaptic inhibition through interneurons. This facilitation may also occur at autonomic ganglia where interneurons have been described.
Because the peripheral nerve responds with only a limited repertoire to the many and varied causes of neuropathy, it is to be expected that other forms of neuropathy and neuralgic hyperpathia (whatever their primary cause) will have many common features. For example, in diabetic neuropathy, the unremitting pain, characteristic cutaneous hypersensitivity, burning sensations, paresthesias, and autonomic symptoms are certainly not specific for diabetes. Histologic findings in nerve biopsy specimens have indicated that the diabetic lesions are predominately in the small fibers, with nerve sprouting (a feature of denervation supersensitivity) the likely cause of the pain.
An enigma in the past, and today a source of great interest to neurobiologists, the importance of denervation supersensitivity with regard to pain has not been appreciated. The implications of Cannon's Law of denervation are probably far more embracing than the few conditions briefly discussed here. It is possible that many other forms of pain, eg, trigeminal or postherpetic (neuralgic) and even chronic low-back pain, are a postdenervation supersensitivity phenomenon rather than the result of noxious stimuli. Thus, pain may be the central perception of 1) an afferent barrage from noxious stimuli or 2) the abnormal input into the central nervous system from ordinarily non-noxious stimuli rendered excessive through overly sensitive receptors (or a variable combination of both). Consider, therefore, the chronic "low back" patient whose discomfort still persists following resolution of the acute phase. Though not crippled or even in distress, he is unable to cope with any but light activities. Such a patient many not be "hyperalgesic" in that ordinarily non-noxious stimuli, eg, prolonged standing, sitting, or walking, can cause symptoms. "Pain" as a scientific term should preferably be discarded and a distinction made between "nociception" and "hyperalgesia," because different approaches are required in their management. A source of nociception should be eliminated-an unstable fracture or spondylolisthesis stabilized, the unrelenting spatial compromise of an impinging disc or carpal tunnel relieved, or the inflammatory and algogenic agents of trauma soothed. In hyperalgesia, any contributory factors from spinal spondylosis should be alleviated (traction, support, mobilization, or even surgery) and the hypersensitive structures desensitized. Lomo has shown in animal experiments that denervation supersensitivity (as assayed by the sensitivity of muscle extrajunctional membrane to acetylcholine) may be reduced or abolished by electrical stimulation. The analgesic effect of transcutaneous neural stimulation may thus depend in part on the reduction of supersensitivity as on the neurohumoral inhibitory effects of the spinal and brainstem antinociceptor systems. Continuous stimulation was found most effective, and it has been suggested that the efficacy of needle acupuncture for hyperalgesia may be due in part to stimulation by the current of injury.
Supersensitivity in autonomic pathways can furthermore lead to the increased blood vessel tone of virtually all tissues and cause secondary pain by structural disintegration. Following denervation, the total collagen in soft and skeletal tissues is reduced. Replacement collagen also has fewer cross-links and is markedly weaker than normal mature collagen. Because collagen provides the strength of ligaments, tendons, cartilage, and bone, this may contribute to many degenerative conditions in the weight-bearing (spinal and intervertebral disc) and activity-stressed parts of the body (tendonitis, cuff tears, epicondylitis, ruptured tendons, and so forth). These secondary conditions, presently dignified by various terms to imply specific clinical entities, are probably only the ultimate sequelae of neuropathy. Degenerative disc disease itself may not be a primary condition. The structural integrity, strength, and reparative capacity of these somatic tissues are such that the constant wear of normal usage is probably adequately compensated for, unless their trophic capability is depressed, as in chronic neuropathy. Thus, in a young person the superaspinatus tendon does not rupture but avulses from its bony insertion, and the intervertebral disc (now thought to be the prime causative factor in spondylosis) is so strong that following violence to the vertebral column, the bones always give way first. The disc is particularly vulnerable to altered vascular tone, being almost avascular and dependent largely upon diffusion through adjacent spongy bone for nutrition. It is food for thought that in all our recent studies, early and subtle signs of peripheral neuropathy were found in a significant number of young (under 30 years), apparently normal, and asymptomatic subjects. Prespondylosis, a term introduced here to describe the early effects of spondylotic attrition on the peripheral nerve, is generally painless, though not necessarily devoid of morbidity. It and its frequent companion, radiculopathy, would therefore seem to be fertile areas for further study in order to understand better the genesis of pain and "degenerative" conditions.
The author is grateful to the Chairman and the Commissioners of the Workers' Compensation Board of British Columbia for their support, and thanks Professor F.P. Patterson, Head, Department of Surgery (Division of Orthopedics), University of British Columbia, Dr. R. M. Feldman, Chairman of the B.C. Section of Physical Medicine and Rehabilitation, and Professor D.D. Murray, Head, Physical Medicine, University of British Columbia, for their held and advice.
© iSTOP - Institute for the Study and Treatment of Pain - 2002